- RÉACTIONNELS (MÉCANISMES)
- RÉACTIONNELS (MÉCANISMES)La loi de Lavoisier permet de définir pour toute réaction chimique un bilan matériel qui est habituellement exprimé sous la forme d’une ou de plusieurs équations de réaction. Un tel bilan ne traduit que d’une façon globale l’ensemble des phénomènes chimiques qui se produisent au niveau moléculaire et qui constituent ce que l’on appelle le mécanisme de la réaction. C’est en chimie moléculaire, et plus particulièrement en chimie organique, que la connaissance des mécanismes réactionnels est la plus avancée; c’est pourquoi on se limitera dans cet article à l’étude des réactions en chimie organique.L’expression d’un mécanisme réactionnel implique l’usage d’un modèle. Les imperfections du modèle de la molécule isolée utilisé à cette fin se retrouvent dans l’expression du modèle mécanistique de la réaction chimique, et les développements de la théorie structurale des molécules ont une répercussion directe sur celui de la représentation des mécanismes de leurs réactions. Les méthodes d’étude des mécanismes font appel aussi bien à l’utilisation des résultats de la cinétique chimique qu’à ceux des méthodes, non cinétiques, d’identification directe des espèces réactionnelles intermédiaires ainsi qu’aux implications indirectes, stéréochimiques notamment, des mécanismes possibles. On peut aujourd’hui décrire et classer l’ensemble des réactions de la chimie organique à partir d’une dizaine de processus élémentaires fondamentaux. Les paramètres sur lesquels repose la classification des réactions sont leur bilan global (substitution, addition, élimination, isomérisation) et le mode de formation ou de rupture des liaisons. Les processus élémentaires, matériaux de base de l’édification des mécanismes, concernent les différents modes d’interaction électronique des réactifs radicalaires intervenant par un électron et des réactifs ioniques ou moléculaires intervenant par un ou plusieurs doublets d’électrons dans l’acte réactionnel. Parmi les réactifs ioniques, on distingue ceux qui présentent une lacune de doublet, et sont électrophiles, de ceux qui disposent d’un doublet, et qui sont nucléophiles. Parmi les centres réactionnels carbonés des substrats, on distingue ceux qui sont tétracoordinés, et qui participent à des réactions de substitution ou d’élimination, de ceux qui sont tri ou dicoordinés, et qui participent à des réactions d’addition. Les réactions font intervenir des intermédiaires, généralement très instables (donc très réactifs), dont les plus importants sont les carbocations, les carbanions, les radicaux libres et les carbènes. La chimie quantique, principalement par la théorie des orbitales moléculaires, permet d’interpréter qualitativement, par des règles simples, l’ensemble des mécanismes connus (recouvrement maximal, conservation de la symétrie, similitude des énergies) et, grâce au développement des méthodes plus élaborées (méthodes ab initio ), il est aujourd’hui effectivement possible d’aborder, sans l’intervention d’aucune donnée empirique autre que celles qui caractérisent les atomes isolés, l’étude théorique quantitative de ces mécanismes réactionnels.1. Définition et élaborationLa réaction chimique d’un substrat organique S (chaîne hydrocarbonée portant des hétéroatomes fonctionnels) avec un réactif R (minéral ou organique) pour donner des produits P, P est décrite par une équation de réaction qui définit son bilan matériel en termes de conservation du nombre et de la nature des éléments qui constituent les différentes espèces en présence (loi de Lavoisier):
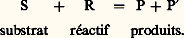 À l’échelle particulaire, ce bilan traduit le résultat global des échanges d’atomes qui ont lieu, au cours de la réaction, entre le substrat et le réactif pour former les nouvelles espèces que sont les produits. Le mécanisme d’une telle réaction est défini comme l’ensemble des processus simples d’échanges d’atomes qui se succèdent entre l’état initial du système (S + R) et son état final (P + P ).La description des mécanismes réactionnels est directement liée à celle des molécules, et leur expression utilise les mêmes modèles moléculaires élaborés par la chimie quantique. À tout progrès réalisé dans la description statique des molécules doit correspondre tôt ou tard un progrès dans la description des mécanismes de leurs réactions; c’est ainsi que, grâce à la mise en œuvre des ordinateurs les plus puissants, on a pu récemment aborder, par les seules méthodes théoriques de la chimie quantique, l’étude de couples de molécules en cours de réaction. Bien que ces tentatives soient actuellement limitées à des cas très simples, il est certain que les progrès réalisés dans la technique du calcul permettront une approche fondamentale et directe des mécanismes réactionnels. Pour l’instant, ces mécanismes ne sont que des schémas pragmatiques faisant intervenir des ouvertures et des fermetures de ce que nos modèles actuels appellent des liaisons et dont le principal mérite est de rendre compréhensibles, et, dans une certaine mesure, prévisibles, les réactions des substrats de la chimie organique.L’élaboration de ces mécanismes repose sur plusieurs types de méthodes. Les méthodes cinétiques (cf. CINÉTIQUE CHIMIQUE) permettent d’établir la loi de vitesse de la réaction et d’atteindre son ordre, relatif aux différents partenaires: on peut ainsi, dans les cas favorables, déterminer la molécularité de l’étape la plus lente du mécanisme; l’étude des effets des isotopes (deutérium) et des solvants sur la cinétique donne également des renseignements structuraux importants sur la nature du complexe activé de l’étape lente. Les méthodes non cinétiques tentent d’identifier directement les espèces intermédiaires de la réaction par une propriété physique observable (spectres de R.P.E., d’absorption en ultraviolet), mais leur mise en œuvre est rendue délicate par la très courte durée de vie autant que par la très faible concentration instantanée de ces espèces. La détermination de la stéréochimie de la réaction permet souvent de préciser la disposition géométrique la plus probable adoptée par l’ensemble du substrat et du réactif au moment de l’acte réactionnel. Enfin, d’une manière générale, on étudie toute modification apportée soit à la cinétique, soit à l’orientation de la réaction, par une perturbation du substrat, du réactif ou des conditions de la réaction.2. Les mécanismes réactionnels, base de classification des réactions de la chimie organiqueProcessus élémentaires des réactionsMalgré leurs imperfections et la simplicité du modèle électronique sur lequel ils reposent, les mécanismes établis pour les réactions les plus diverses de la chimie organique ont permis de réduire à un nombre relativement restreint les processus élémentaires fondamentaux selon lesquels il est aujourd’hui possible de décrire et de classer l’ensemble de ces réactions. Une telle classification repose sur le choix de deux paramètres, qui sont le bilan global de la réaction (substitution, addition, élimination, isomérisation), et le mode de formation (ou mode d’ouverture) de la liaison qui s’établit (ou se rompt), au cours de la réaction, entre le réactif et le substrat (processus mono-ou biélectronique, c’est-à-dire radicalaire ou ionique, ou éventuellement polyélectronique, c’est-à-dire moléculaire).Les mécanismes réactionnels sont rarement simples et correspondent le plus souvent à une succession d’étapes élémentaires qui représentent réellement la base commune à tous les mécanismes. Ces processus élémentaires font intervenir, dans une étape généralement bimoléculaire, un réactif qui apporte l’amorce de la nouvelle liaison qui va se former et un substrat qui contient le centre , généralement carboné, autour duquel se produit l’échange d’électrons, objet de la réaction.On distingue les réactifs radicalaires , qui contribuent par un nombre impair d’électrons à la formation des nouvelles liaisons, et les réactifs ioniques ou moléculaires , qui y contribuent par un nombre pair d’électrons: réactifs électrophiles , présentant une lacune de doublet (orbitale vide, actuelle ou potentielle), réactifs nucléophiles , présentant un doublet disponible, réactifs moléculaires , présentant plusieurs doublets (généralement 神 dans un système conjugué) simultanément disponibles pour constituer plusieurs liaisons nouvelles.On distingue d’autre part les centres saturés , pour lesquels l’atome central possédant son maximum de coordination (4 pour les atomes de la seconde ligne de la classification, notamment pour le carbone) est engagé dans la formation de liaisons de type exclusivement 靖, et les centres insaturés où l’atome central échange un ou plusieurs doublets 神 avec le reste du squelette.De la combinaison de ces deux paramètres résulte la définition des principaux processus simples des mécanismes réactionnels en chimie organique, tels qu’ils apparaissent dans le tableau 1:(1) Processus homolytique d’arrachement d’un atome (généralement H.) sur un centre réactionnel saturé du substrat et qui le transforme en radical libre (processus de transfert du radical);(2) Processus homolytique de combinaison de deux radicaux libres, inverse de la réaction d’homolyse des liaisons;(3) Processus homolytique unimoléculaire de fragmentation d’un radical libre, conduisant à un autre radical, plus simple, et à une molécule stable;(4) Processus homolytique d’addition d’un réactif radicalaire sur un substrat insaturé avec formation d’un nouveau radical libre;(5) Processus homolytique de dismutation de deux radicaux pour donner deux molécules stables (dont une insaturée), processus qui correspond à la superposition d’un transfert et d’une fragmentation;(6) Processus hétérolytique de remplacement d’un groupe par un autre, apporté par le réactif, sur un centre réactionnel saturé du substrat, le réactif pouvant être nucléophile (6 a) ou électrophile (6 b);(7) Processus hétérolytique d’addition d’un réactif ionique sur un substrat insaturé, le réactif pouvant être nucléophile (7 a) ou électrophile (7 b);(8) Processus hétérolytique d’arrachement d’un atome ou d’un groupe, conduisant à un produit insaturé, sous l’action d’un réactif basique B size=1漣 (8 a) ou acide A+ (8 b);(9) Processus moléculaire (concerté) d’addition de deux substrats insaturés, comme dans la cycloaddition de Diels-Alder.Ces processus élémentaires, éventuellement combinés dans des étapes successives, constituent les éléments fondamentaux des mécanismes réactionnels en chimie organique.Principaux intermédiaires des réactionsLe déroulement de la réaction à l’échelle moléculaire [cf. CINÉTIQUE CHIMIQUE] fait intervenir, pour chacune des étapes élémentaires, une forme intermédiaire active qui, en fonction de sa durée de vie ( 精), est soit un état de transition ( 精 = 10 size=1漣13 s), soit une espèce intermédiaire ( 精 閭 10 size=1漣12 s). Dans ce dernier cas, il a été possible d’établir certaines caractéristiques structurales et réactionnelles des principaux intermédiaires, soit indirectement (conséquences cinétiques), soit directement (spectroscopies), qui sont les carbocations, les carbanions, les radicaux libres, les carbènes, les nitrènes et les arynes.CarbocationsLes carbocations, espèces intermédiaires cationiques construites autour d’un atome de carbone tricoordiné, dans l’état électronique sp 2 (cf. figure, en a ), présentent une lacune de doublet (orbitale disponible) et sont donc positivement chargés; ils sont obtenus comme intermédiaires dans diverses réactions hétérolytiques (substitutions, additions). Très réactifs vis-à-vis des espèces nucléophiles (substitution, addition) et basiques (élimination), ils sont caractérisés par une aptitude marquée au réarrangement, lorsque ce dernier les transforme en carbocations plus stables. Cette augmentation de la stabilité peut résulter soit d’une décompression stérique plus importante du centre sp 3 (qui devient cationique sp 2) par suite d’une augmentation du degré de substitution de ce centre, soit d’une augmentation de la dispersion de charge positive du carbocation, par conjugaison du centre cationique avec un système 神 par exemple; ils ne sont éventuellement stables qu’en milieu acide (solvatation).Le tableau 2 donne quelques exemples de formation d’intermédiaires carbocationiques:(10) Hétérolyse thermique d’un sel d’alkyldiazonium;(11) Addition d’un acide halogéné sur une oléfine ramifiée;(12) Hétérolyse du perchlorate d’acétylium;(13) Hétérolyse du sulfate de triphénylméthyle.Les carbocations formés, de structure sp 2 (cf. figure, en a ), se classent dans l’ordre suivant de stabilité croissante:
À l’échelle particulaire, ce bilan traduit le résultat global des échanges d’atomes qui ont lieu, au cours de la réaction, entre le substrat et le réactif pour former les nouvelles espèces que sont les produits. Le mécanisme d’une telle réaction est défini comme l’ensemble des processus simples d’échanges d’atomes qui se succèdent entre l’état initial du système (S + R) et son état final (P + P ).La description des mécanismes réactionnels est directement liée à celle des molécules, et leur expression utilise les mêmes modèles moléculaires élaborés par la chimie quantique. À tout progrès réalisé dans la description statique des molécules doit correspondre tôt ou tard un progrès dans la description des mécanismes de leurs réactions; c’est ainsi que, grâce à la mise en œuvre des ordinateurs les plus puissants, on a pu récemment aborder, par les seules méthodes théoriques de la chimie quantique, l’étude de couples de molécules en cours de réaction. Bien que ces tentatives soient actuellement limitées à des cas très simples, il est certain que les progrès réalisés dans la technique du calcul permettront une approche fondamentale et directe des mécanismes réactionnels. Pour l’instant, ces mécanismes ne sont que des schémas pragmatiques faisant intervenir des ouvertures et des fermetures de ce que nos modèles actuels appellent des liaisons et dont le principal mérite est de rendre compréhensibles, et, dans une certaine mesure, prévisibles, les réactions des substrats de la chimie organique.L’élaboration de ces mécanismes repose sur plusieurs types de méthodes. Les méthodes cinétiques (cf. CINÉTIQUE CHIMIQUE) permettent d’établir la loi de vitesse de la réaction et d’atteindre son ordre, relatif aux différents partenaires: on peut ainsi, dans les cas favorables, déterminer la molécularité de l’étape la plus lente du mécanisme; l’étude des effets des isotopes (deutérium) et des solvants sur la cinétique donne également des renseignements structuraux importants sur la nature du complexe activé de l’étape lente. Les méthodes non cinétiques tentent d’identifier directement les espèces intermédiaires de la réaction par une propriété physique observable (spectres de R.P.E., d’absorption en ultraviolet), mais leur mise en œuvre est rendue délicate par la très courte durée de vie autant que par la très faible concentration instantanée de ces espèces. La détermination de la stéréochimie de la réaction permet souvent de préciser la disposition géométrique la plus probable adoptée par l’ensemble du substrat et du réactif au moment de l’acte réactionnel. Enfin, d’une manière générale, on étudie toute modification apportée soit à la cinétique, soit à l’orientation de la réaction, par une perturbation du substrat, du réactif ou des conditions de la réaction.2. Les mécanismes réactionnels, base de classification des réactions de la chimie organiqueProcessus élémentaires des réactionsMalgré leurs imperfections et la simplicité du modèle électronique sur lequel ils reposent, les mécanismes établis pour les réactions les plus diverses de la chimie organique ont permis de réduire à un nombre relativement restreint les processus élémentaires fondamentaux selon lesquels il est aujourd’hui possible de décrire et de classer l’ensemble de ces réactions. Une telle classification repose sur le choix de deux paramètres, qui sont le bilan global de la réaction (substitution, addition, élimination, isomérisation), et le mode de formation (ou mode d’ouverture) de la liaison qui s’établit (ou se rompt), au cours de la réaction, entre le réactif et le substrat (processus mono-ou biélectronique, c’est-à-dire radicalaire ou ionique, ou éventuellement polyélectronique, c’est-à-dire moléculaire).Les mécanismes réactionnels sont rarement simples et correspondent le plus souvent à une succession d’étapes élémentaires qui représentent réellement la base commune à tous les mécanismes. Ces processus élémentaires font intervenir, dans une étape généralement bimoléculaire, un réactif qui apporte l’amorce de la nouvelle liaison qui va se former et un substrat qui contient le centre , généralement carboné, autour duquel se produit l’échange d’électrons, objet de la réaction.On distingue les réactifs radicalaires , qui contribuent par un nombre impair d’électrons à la formation des nouvelles liaisons, et les réactifs ioniques ou moléculaires , qui y contribuent par un nombre pair d’électrons: réactifs électrophiles , présentant une lacune de doublet (orbitale vide, actuelle ou potentielle), réactifs nucléophiles , présentant un doublet disponible, réactifs moléculaires , présentant plusieurs doublets (généralement 神 dans un système conjugué) simultanément disponibles pour constituer plusieurs liaisons nouvelles.On distingue d’autre part les centres saturés , pour lesquels l’atome central possédant son maximum de coordination (4 pour les atomes de la seconde ligne de la classification, notamment pour le carbone) est engagé dans la formation de liaisons de type exclusivement 靖, et les centres insaturés où l’atome central échange un ou plusieurs doublets 神 avec le reste du squelette.De la combinaison de ces deux paramètres résulte la définition des principaux processus simples des mécanismes réactionnels en chimie organique, tels qu’ils apparaissent dans le tableau 1:(1) Processus homolytique d’arrachement d’un atome (généralement H.) sur un centre réactionnel saturé du substrat et qui le transforme en radical libre (processus de transfert du radical);(2) Processus homolytique de combinaison de deux radicaux libres, inverse de la réaction d’homolyse des liaisons;(3) Processus homolytique unimoléculaire de fragmentation d’un radical libre, conduisant à un autre radical, plus simple, et à une molécule stable;(4) Processus homolytique d’addition d’un réactif radicalaire sur un substrat insaturé avec formation d’un nouveau radical libre;(5) Processus homolytique de dismutation de deux radicaux pour donner deux molécules stables (dont une insaturée), processus qui correspond à la superposition d’un transfert et d’une fragmentation;(6) Processus hétérolytique de remplacement d’un groupe par un autre, apporté par le réactif, sur un centre réactionnel saturé du substrat, le réactif pouvant être nucléophile (6 a) ou électrophile (6 b);(7) Processus hétérolytique d’addition d’un réactif ionique sur un substrat insaturé, le réactif pouvant être nucléophile (7 a) ou électrophile (7 b);(8) Processus hétérolytique d’arrachement d’un atome ou d’un groupe, conduisant à un produit insaturé, sous l’action d’un réactif basique B size=1漣 (8 a) ou acide A+ (8 b);(9) Processus moléculaire (concerté) d’addition de deux substrats insaturés, comme dans la cycloaddition de Diels-Alder.Ces processus élémentaires, éventuellement combinés dans des étapes successives, constituent les éléments fondamentaux des mécanismes réactionnels en chimie organique.Principaux intermédiaires des réactionsLe déroulement de la réaction à l’échelle moléculaire [cf. CINÉTIQUE CHIMIQUE] fait intervenir, pour chacune des étapes élémentaires, une forme intermédiaire active qui, en fonction de sa durée de vie ( 精), est soit un état de transition ( 精 = 10 size=1漣13 s), soit une espèce intermédiaire ( 精 閭 10 size=1漣12 s). Dans ce dernier cas, il a été possible d’établir certaines caractéristiques structurales et réactionnelles des principaux intermédiaires, soit indirectement (conséquences cinétiques), soit directement (spectroscopies), qui sont les carbocations, les carbanions, les radicaux libres, les carbènes, les nitrènes et les arynes.CarbocationsLes carbocations, espèces intermédiaires cationiques construites autour d’un atome de carbone tricoordiné, dans l’état électronique sp 2 (cf. figure, en a ), présentent une lacune de doublet (orbitale disponible) et sont donc positivement chargés; ils sont obtenus comme intermédiaires dans diverses réactions hétérolytiques (substitutions, additions). Très réactifs vis-à-vis des espèces nucléophiles (substitution, addition) et basiques (élimination), ils sont caractérisés par une aptitude marquée au réarrangement, lorsque ce dernier les transforme en carbocations plus stables. Cette augmentation de la stabilité peut résulter soit d’une décompression stérique plus importante du centre sp 3 (qui devient cationique sp 2) par suite d’une augmentation du degré de substitution de ce centre, soit d’une augmentation de la dispersion de charge positive du carbocation, par conjugaison du centre cationique avec un système 神 par exemple; ils ne sont éventuellement stables qu’en milieu acide (solvatation).Le tableau 2 donne quelques exemples de formation d’intermédiaires carbocationiques:(10) Hétérolyse thermique d’un sel d’alkyldiazonium;(11) Addition d’un acide halogéné sur une oléfine ramifiée;(12) Hétérolyse du perchlorate d’acétylium;(13) Hétérolyse du sulfate de triphénylméthyle.Les carbocations formés, de structure sp 2 (cf. figure, en a ), se classent dans l’ordre suivant de stabilité croissante: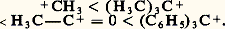 CarbanionsEspèces intermédiaires anioniques construites autour d’un atome de carbone tricoordiné, dans l’état électronique sp 3 (cf. figure, en b ), les carbanions présentent un doublet non partagé et sont chargés négativement; ils sont obtenus comme intermédiaires dans diverses réactions hétérolytiques (substitutions, éliminations) et sont très réactifs vis-à-vis des espèces électrophiles (substitutions, additions) et acides (élimination). Leur stabilisation est essentiellement assurée par la dispersion de leur charge négative (par effet induit ou par conjugaison du centre anionique avec un système 神 par exemple); ils ne sont éventuellement stables qu’en milieu basique (solvatation).Des exemples de formation d’intermédiaires carbanioniques sont donnés dans le tableau 3:(14) Hétérolyse d’un organométallique avec l’hexaméthylphosphotriamide (HMPT) comme catalyseur;(15) Arrachement par une base d’un proton acide du substrat;(16) Décarboxylation d’un acide activé par un groupement stabilisateur du carbanion.Radicaux libresEspèces intermédiaires neutres construites autour d’un carbone tricoordiné, dans l’état électronique sp 2 (cf. figure, en c ), les radicaux libres contiennent un nombre impair d’électrons, c’est-à-dire qu’ils possèdent un électron non apparié (électron solitaire); ils sont obtenus comme intermédiaires dans de nombreuses réactions homolytiques, fréquemment en chaîne [cf. CINÉTIQUE CHIMIQUE], soit par rupture thermique d’une liaison (thermolyse), soit par rupture photochimique (photolyse), soit par arrachement d’un atome d’hydrogène (initiation par un générateur de radicaux libres), soit par réduction (monoélectronique) d’un carbocation ou oxydation (monoélectronique) d’un carbanion.Des exemples de formation d’intermédiaires radicalaires figurent dans le tableau 4:(17) Thermolyse d’une molécule présentant une liaison fragile;(18) Photolyse d’une molécule présentant, dans son état excité, une liaison fragile;(19) Arrachement d’un atome d’hydrogène par un réactif radicalaire;(20) Réduction d’un carbocation par un réducteur monoélectronique.À la différence des carbocations et des carbanions, les radicaux libres sont peu solvatés et présentent généralement une réactivité considérable, soit dans des réactions de combinaison avec un autre radical libre, soit dans des processus d’arrachement ou de fragmentation.CarbènesLes carbènes sont des espèces intermédiaires neutres du carbone bicoordiné, pouvant présenter, dans leur état singulet, la structure à la fois d’un carbanion et d’un carbocation construit autour d’un atome de carbone dans l’état sp 2 (cf. figure, en d ), et, dans leur état triplet, celle d’un biradical construit autour d’un atome de carbone sp 3 (cf. figure, en e ); ils sont observés comme intermédiaires dans des processus thermo- ou photolytiques particuliers (diazo-alcanes, cétène) ou dans des réactions de déshalogénation, en milieu basique, de dérivés gem trihalogénés. Ils interviennent dans des réactions d’insertion et de cis -addition.Le tableau 5 donne quelques exemples de formation de carbènes intermédiaires:(21) Photolyse du cétène (21 a) suivie d’une insertion sur un hydrocarbure saturé (21 b);(22) Photolyse du diazométhane (22 a) suivie d’une cis -addition sur une oléfine (22 b);(23) Élimination par une base de HCl sur le chloroforme (23 a) suivie de l’addition sur un phénate (23 b, réaction de Reimer et Tiemann).Autres intermédiairesLes nitrènes sont des espèces intermédiaires neutres de l’azote monocoordiné, structuralement analogues aux carbènes et obtenues par photolyse ou thermolyse des azides (tabl. 6, réaction 24).Les arynes sont des espèces intermédiaires neutres comportant deux carbones adjacents bicoordinés, inclus dans une structure aromatique, pouvant présenter, dans un état singulet, la structure d’un orthocarbocation-carbanion et, dans un état triplet, celle d’un ortho-bis radical libre. Ils interviennent dans des réactions d’élimination-addition concernant certains substrats aromatiques. Il y a formation d’arynes intermédiaires par arrachement par une base forte d’une molécule d’acide chlorhydrique à un chlorure d’aryle (25 a); elle est suivie d’une addition (25 b).D’une manière générale, les molécules peuvent accroître leur réactivité ou transformer leur mode de réaction en modifiant leur structure électronique (molécules activées ), soit par absorption d’une radiation d’énergie convenable (activation photochimique), soit par formation d’un adduit avec un catalyseur (activation catalytique).Tous ces intermédiaires, principalement lorsqu’ils portent une charge électrique, sont solvatés dans le milieu où ils se forment et réagissent.3. Aspects quanto-chimiques des mécanismes réactionnelsL’aspect cinétique des mécanismes ayant été déjà envisagé dans l’article CINÉTIQUE CHIMIQUE, il est utile de préciser ici les résultats de leur étude sous l’angle de la mécanique quantique. La mécanique ondulatoire ne permet, pour un certain nombre d’années encore, que de proposer un modèle très qualitatif pour la réaction chimique à l’échelle des molécules, disons pour l’acte chimique. Ce modèle utilise celui, déjà bien élaboré, de la molécule isolée qu’il déforme par des perturbations directement inspirées par le modèle cinétique. Pour l’ensemble substrat + réactif, l’énergie est évaluée en fonction de la distribution spatiale des atomes (surfaces équipotentielles), et les chemins réactionnels les plus probables sont obtenus par minimisation de l’énergie du système global (vallées des surfaces de potentiel). Cette approche théorique ab initio n’en est qu’à ses débuts et il serait illusoire d’en attendre momentanément autre chose qu’un modèle provisoire et très qualitatif de l’acte réactionnel.De leur côté, les méthodes simplifiées de la chimie quantique abordent le problème de la réactivité de deux manières différentes: par le modèle de la molécule isolée, on recherche la corrélation entre réactivité et caractéristiques statiques (indices structuraux) du seul substrat; par le modèle du complexe intermédiaire, on recherche la corrélation entre réactivité et énergie du complexe intermédiaire supposé (énergies de polarisation du substrat).Ces deux modèles sont très grossiers et ne peuvent être utilisés, à la limite, que dans la comparaison de plusieurs substrats d’une même famille ou de plusieurs centres réactionnels d’un même substrat (hypothèse de la constance de l’entropie d’activation dans la série des réactions étudiées). Sur un plan encore plus qualitatif, notons un certain nombre de règles utiles qui découlent de ces études théoriques:Règle du recouvrement maximal : l’énergie de l’état de transition est d’autant plus petite que le recouvrement des orbitales impliquées dans l’acte réactionnel est plus important (orientation et stéréochimie des réactions);Règles de symétrie des réactions : les conditions de symétrie dans lesquelles se produisent les réactions doivent se refléter au niveau des orbitales impliquées dans l’acte réactionnel (règles dites de Woodward et Hoffmann);Règle de similitude des énergies : l’énergie de l’état de transition est d’autant plus petite que les énergies des électrons impliqués et que les polarisabilités (énergie de déformation) des orbitales qui les décrivent sont respectivement plus voisines.4. Exemples typiques de mécanismes réactionnelsLa substitution nucléophile sur un centre saturéLe bilan de la substitution nucléophile sur un centre saturé s’écrit:
CarbanionsEspèces intermédiaires anioniques construites autour d’un atome de carbone tricoordiné, dans l’état électronique sp 3 (cf. figure, en b ), les carbanions présentent un doublet non partagé et sont chargés négativement; ils sont obtenus comme intermédiaires dans diverses réactions hétérolytiques (substitutions, éliminations) et sont très réactifs vis-à-vis des espèces électrophiles (substitutions, additions) et acides (élimination). Leur stabilisation est essentiellement assurée par la dispersion de leur charge négative (par effet induit ou par conjugaison du centre anionique avec un système 神 par exemple); ils ne sont éventuellement stables qu’en milieu basique (solvatation).Des exemples de formation d’intermédiaires carbanioniques sont donnés dans le tableau 3:(14) Hétérolyse d’un organométallique avec l’hexaméthylphosphotriamide (HMPT) comme catalyseur;(15) Arrachement par une base d’un proton acide du substrat;(16) Décarboxylation d’un acide activé par un groupement stabilisateur du carbanion.Radicaux libresEspèces intermédiaires neutres construites autour d’un carbone tricoordiné, dans l’état électronique sp 2 (cf. figure, en c ), les radicaux libres contiennent un nombre impair d’électrons, c’est-à-dire qu’ils possèdent un électron non apparié (électron solitaire); ils sont obtenus comme intermédiaires dans de nombreuses réactions homolytiques, fréquemment en chaîne [cf. CINÉTIQUE CHIMIQUE], soit par rupture thermique d’une liaison (thermolyse), soit par rupture photochimique (photolyse), soit par arrachement d’un atome d’hydrogène (initiation par un générateur de radicaux libres), soit par réduction (monoélectronique) d’un carbocation ou oxydation (monoélectronique) d’un carbanion.Des exemples de formation d’intermédiaires radicalaires figurent dans le tableau 4:(17) Thermolyse d’une molécule présentant une liaison fragile;(18) Photolyse d’une molécule présentant, dans son état excité, une liaison fragile;(19) Arrachement d’un atome d’hydrogène par un réactif radicalaire;(20) Réduction d’un carbocation par un réducteur monoélectronique.À la différence des carbocations et des carbanions, les radicaux libres sont peu solvatés et présentent généralement une réactivité considérable, soit dans des réactions de combinaison avec un autre radical libre, soit dans des processus d’arrachement ou de fragmentation.CarbènesLes carbènes sont des espèces intermédiaires neutres du carbone bicoordiné, pouvant présenter, dans leur état singulet, la structure à la fois d’un carbanion et d’un carbocation construit autour d’un atome de carbone dans l’état sp 2 (cf. figure, en d ), et, dans leur état triplet, celle d’un biradical construit autour d’un atome de carbone sp 3 (cf. figure, en e ); ils sont observés comme intermédiaires dans des processus thermo- ou photolytiques particuliers (diazo-alcanes, cétène) ou dans des réactions de déshalogénation, en milieu basique, de dérivés gem trihalogénés. Ils interviennent dans des réactions d’insertion et de cis -addition.Le tableau 5 donne quelques exemples de formation de carbènes intermédiaires:(21) Photolyse du cétène (21 a) suivie d’une insertion sur un hydrocarbure saturé (21 b);(22) Photolyse du diazométhane (22 a) suivie d’une cis -addition sur une oléfine (22 b);(23) Élimination par une base de HCl sur le chloroforme (23 a) suivie de l’addition sur un phénate (23 b, réaction de Reimer et Tiemann).Autres intermédiairesLes nitrènes sont des espèces intermédiaires neutres de l’azote monocoordiné, structuralement analogues aux carbènes et obtenues par photolyse ou thermolyse des azides (tabl. 6, réaction 24).Les arynes sont des espèces intermédiaires neutres comportant deux carbones adjacents bicoordinés, inclus dans une structure aromatique, pouvant présenter, dans un état singulet, la structure d’un orthocarbocation-carbanion et, dans un état triplet, celle d’un ortho-bis radical libre. Ils interviennent dans des réactions d’élimination-addition concernant certains substrats aromatiques. Il y a formation d’arynes intermédiaires par arrachement par une base forte d’une molécule d’acide chlorhydrique à un chlorure d’aryle (25 a); elle est suivie d’une addition (25 b).D’une manière générale, les molécules peuvent accroître leur réactivité ou transformer leur mode de réaction en modifiant leur structure électronique (molécules activées ), soit par absorption d’une radiation d’énergie convenable (activation photochimique), soit par formation d’un adduit avec un catalyseur (activation catalytique).Tous ces intermédiaires, principalement lorsqu’ils portent une charge électrique, sont solvatés dans le milieu où ils se forment et réagissent.3. Aspects quanto-chimiques des mécanismes réactionnelsL’aspect cinétique des mécanismes ayant été déjà envisagé dans l’article CINÉTIQUE CHIMIQUE, il est utile de préciser ici les résultats de leur étude sous l’angle de la mécanique quantique. La mécanique ondulatoire ne permet, pour un certain nombre d’années encore, que de proposer un modèle très qualitatif pour la réaction chimique à l’échelle des molécules, disons pour l’acte chimique. Ce modèle utilise celui, déjà bien élaboré, de la molécule isolée qu’il déforme par des perturbations directement inspirées par le modèle cinétique. Pour l’ensemble substrat + réactif, l’énergie est évaluée en fonction de la distribution spatiale des atomes (surfaces équipotentielles), et les chemins réactionnels les plus probables sont obtenus par minimisation de l’énergie du système global (vallées des surfaces de potentiel). Cette approche théorique ab initio n’en est qu’à ses débuts et il serait illusoire d’en attendre momentanément autre chose qu’un modèle provisoire et très qualitatif de l’acte réactionnel.De leur côté, les méthodes simplifiées de la chimie quantique abordent le problème de la réactivité de deux manières différentes: par le modèle de la molécule isolée, on recherche la corrélation entre réactivité et caractéristiques statiques (indices structuraux) du seul substrat; par le modèle du complexe intermédiaire, on recherche la corrélation entre réactivité et énergie du complexe intermédiaire supposé (énergies de polarisation du substrat).Ces deux modèles sont très grossiers et ne peuvent être utilisés, à la limite, que dans la comparaison de plusieurs substrats d’une même famille ou de plusieurs centres réactionnels d’un même substrat (hypothèse de la constance de l’entropie d’activation dans la série des réactions étudiées). Sur un plan encore plus qualitatif, notons un certain nombre de règles utiles qui découlent de ces études théoriques:Règle du recouvrement maximal : l’énergie de l’état de transition est d’autant plus petite que le recouvrement des orbitales impliquées dans l’acte réactionnel est plus important (orientation et stéréochimie des réactions);Règles de symétrie des réactions : les conditions de symétrie dans lesquelles se produisent les réactions doivent se refléter au niveau des orbitales impliquées dans l’acte réactionnel (règles dites de Woodward et Hoffmann);Règle de similitude des énergies : l’énergie de l’état de transition est d’autant plus petite que les énergies des électrons impliqués et que les polarisabilités (énergie de déformation) des orbitales qui les décrivent sont respectivement plus voisines.4. Exemples typiques de mécanismes réactionnelsLa substitution nucléophile sur un centre saturéLe bilan de la substitution nucléophile sur un centre saturé s’écrit: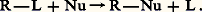 Deux mécanismes principaux peuvent intervenir selon la nature du groupe partant (face=F0019 漣 L), l’état de contrainte (stérique) existant autour du carbone qui est le centre réactionnel, la nature du nucléophile (charge, polarisabilité, encombrement autour du centre nucléophile), le solvant, la température, la présence éventuelle d’autres réactifs (sels, ions communs), etc.Dans leur principe, ces deux mécanismes sont schématisés de la manière suivante (tabl. 7):Le mécanisme «bimoléculaire» , S2, procède en une étape unique (26), au cours de laquelle le réactif nucléophile Nu remplace progressivement le groupe partant L. La stéréochimie de cette réaction est conforme au principe de recouvrement maximal: l’orbitale moléculaire du doublet apporté par le nucléophile recouvre au maximum l’orbitale moléculaire antiliante 靖C 漣 L du substrat si l’approche est longitudinale dans l’axe de cette orbitale 靖; l’attaque est dorsale et la réaction est normalement accompagnée d’une inversion de configuration au niveau du centre réactionnel carboné (inversion de Walden ). L’état de transition supposé met en évidence ce recouvrement maximal (27).Le mécanisme «unimoléculaire» , S1, procède en deux étapes: la première (28 a), qui est la plus lente (étape cinétique), correspond à une hétérolyse, sous l’action du solvant S, de la liaison C 漣 L pour former un intermédiaire carbocationique; la seconde (28 b) est l’attaque rapide de ce carbocation par le réactif nucléophile.Le rôle du solvant est essentiel, car l’énergie mise en jeu dans la séparation de charges qui accompagne l’hétérolyse de la liaison C 漣 L peut être compensée par la stabilisation apportée par la solvatation des ions formés. Pour justifier l’ensemble des résultats cinétiques observés, il est nécessaire de faire intervenir plusieurs étapes dans le processus d’hétérolyse (29). La stéréochimie de la réaction de solvolyse (le solvant est en même temps le nucléophile) correspond bien au principe de recouvrement maximal: l’attaque est orthogonale au plan nodal du carbocation, mais elle est dorsale (inversion) ou indifféremment dorsale ou frontale (racémisation) selon que la deuxième étape de la réaction a lieu sur le carbocation dissymétriquement solvaté (avant dissociation) ou symétriquement solvaté (après dissociation).Il est fréquent que les deux mécanismes S1 et S2 interviennent parallèlement pour une réaction; cependant, le mécanisme S1 est principalement observé lorsque le substrat est susceptible de donner un carbocation (décompression stérique par hétérolyse, dispersion de la charge positive par effet inductif ou par conjugaison), lorsque le réactif est faiblement nucléophile, le solvant présentant un bon pouvoir ionisant.L’élimination sur un substrat saturéLe bilan d’une élimination sur un substrat saturé correspond au remplacement, par une liaison 神, de deux liaisons 靖. Cette réaction est généralement provoquée par une base, et l’un des groupes éliminés par cette base est un proton (30).Comme pour la substitution nucléophile, deux mécanismes principaux peuvent intervenir selon la plus ou moins grande facilité de formation d’un carbocation intermédiaire: Le mécanisme «bimoléculaire» , E2, procède en une seule étape (31), au cours de laquelle le réactif basique arrache progressivement le proton, tandis que le groupe partant L s’éloigne du substrat. La stéréochimie de cette réaction est conforme au principe de recouvrement maximal, car dans le complexe activé, les orbitales 靖 impliquées ont leurs axes alignés ou parallèles (32). L’attaque est normalement antiparallèle; le produit obtenu correspond à une élimination trans de H+ et de L size=1漣.Le mécanisme «unimoléculaire» , E1, procède en deux étapes: la première (33 a), l’étape cinétique, est une hétérolyse de la liaison C 漣 L, identique à la première étape du mécanisme S1; le carbocation formé subit, sous l’action de la base, l’arrachement du proton (33 b). Cette réaction, parallèle à la réaction S1, accompagne normalement cette dernière comme réaction secondaire.Lorsque plusieurs protons sont disponibles sur des atomes de carbone différents situés en 見 du centre réactionnel, l’orientation de la réaction dépend de facteurs stériques (34).La substitution électrophile sur substrat aromatique procède de mécanismes qui correspondent à la succession d’une addition électrophile sur le substrat aromatique et d’une élimination sur l’intermédiaire obtenu.
Deux mécanismes principaux peuvent intervenir selon la nature du groupe partant (face=F0019 漣 L), l’état de contrainte (stérique) existant autour du carbone qui est le centre réactionnel, la nature du nucléophile (charge, polarisabilité, encombrement autour du centre nucléophile), le solvant, la température, la présence éventuelle d’autres réactifs (sels, ions communs), etc.Dans leur principe, ces deux mécanismes sont schématisés de la manière suivante (tabl. 7):Le mécanisme «bimoléculaire» , S2, procède en une étape unique (26), au cours de laquelle le réactif nucléophile Nu remplace progressivement le groupe partant L. La stéréochimie de cette réaction est conforme au principe de recouvrement maximal: l’orbitale moléculaire du doublet apporté par le nucléophile recouvre au maximum l’orbitale moléculaire antiliante 靖C 漣 L du substrat si l’approche est longitudinale dans l’axe de cette orbitale 靖; l’attaque est dorsale et la réaction est normalement accompagnée d’une inversion de configuration au niveau du centre réactionnel carboné (inversion de Walden ). L’état de transition supposé met en évidence ce recouvrement maximal (27).Le mécanisme «unimoléculaire» , S1, procède en deux étapes: la première (28 a), qui est la plus lente (étape cinétique), correspond à une hétérolyse, sous l’action du solvant S, de la liaison C 漣 L pour former un intermédiaire carbocationique; la seconde (28 b) est l’attaque rapide de ce carbocation par le réactif nucléophile.Le rôle du solvant est essentiel, car l’énergie mise en jeu dans la séparation de charges qui accompagne l’hétérolyse de la liaison C 漣 L peut être compensée par la stabilisation apportée par la solvatation des ions formés. Pour justifier l’ensemble des résultats cinétiques observés, il est nécessaire de faire intervenir plusieurs étapes dans le processus d’hétérolyse (29). La stéréochimie de la réaction de solvolyse (le solvant est en même temps le nucléophile) correspond bien au principe de recouvrement maximal: l’attaque est orthogonale au plan nodal du carbocation, mais elle est dorsale (inversion) ou indifféremment dorsale ou frontale (racémisation) selon que la deuxième étape de la réaction a lieu sur le carbocation dissymétriquement solvaté (avant dissociation) ou symétriquement solvaté (après dissociation).Il est fréquent que les deux mécanismes S1 et S2 interviennent parallèlement pour une réaction; cependant, le mécanisme S1 est principalement observé lorsque le substrat est susceptible de donner un carbocation (décompression stérique par hétérolyse, dispersion de la charge positive par effet inductif ou par conjugaison), lorsque le réactif est faiblement nucléophile, le solvant présentant un bon pouvoir ionisant.L’élimination sur un substrat saturéLe bilan d’une élimination sur un substrat saturé correspond au remplacement, par une liaison 神, de deux liaisons 靖. Cette réaction est généralement provoquée par une base, et l’un des groupes éliminés par cette base est un proton (30).Comme pour la substitution nucléophile, deux mécanismes principaux peuvent intervenir selon la plus ou moins grande facilité de formation d’un carbocation intermédiaire: Le mécanisme «bimoléculaire» , E2, procède en une seule étape (31), au cours de laquelle le réactif basique arrache progressivement le proton, tandis que le groupe partant L s’éloigne du substrat. La stéréochimie de cette réaction est conforme au principe de recouvrement maximal, car dans le complexe activé, les orbitales 靖 impliquées ont leurs axes alignés ou parallèles (32). L’attaque est normalement antiparallèle; le produit obtenu correspond à une élimination trans de H+ et de L size=1漣.Le mécanisme «unimoléculaire» , E1, procède en deux étapes: la première (33 a), l’étape cinétique, est une hétérolyse de la liaison C 漣 L, identique à la première étape du mécanisme S1; le carbocation formé subit, sous l’action de la base, l’arrachement du proton (33 b). Cette réaction, parallèle à la réaction S1, accompagne normalement cette dernière comme réaction secondaire.Lorsque plusieurs protons sont disponibles sur des atomes de carbone différents situés en 見 du centre réactionnel, l’orientation de la réaction dépend de facteurs stériques (34).La substitution électrophile sur substrat aromatique procède de mécanismes qui correspondent à la succession d’une addition électrophile sur le substrat aromatique et d’une élimination sur l’intermédiaire obtenu.
Encyclopédie Universelle. 2012.